pE (måttenhet)
Från Rilpedia

pE är ett logaritmiskt mått på elektronaktiviteten i en kemisk lösning (t ex markvätska). Höga pE-värden indikerar oxiderande förhållanden med god tillgång på syrgas och låg elektronaktivitet. Låga pE-värden indikerar reducerande förhållanden, med frånvaro av syrgas (syrgasbrist) och hög elektronaktivitet.
Strikt matematiskt definieras pE som den negativa 10-logaritmen av elektronaktiviteten, d v s pE = -log10(e-) där (e-) är elektonaktiviteten. pE är matematiskt kopplad till elektrodpotentialen (EH) genom sambandet EH = 0,05916 pE.
Innehåll |
Olika redoxförhållanden
Inom markkemin brukar man skilja på oxiska, suboxiska och anoxiska förhållanden. Vid oxiska förhållanden är pE över 7 och syrgas finns att tillgå för biokemiska reaktioner. Vid suboxiska förhållanden är pE 2-7 har syrgasen tagit slut. Vid anoxiska förhållanden är pE lägre än +2. Dessa gränsvärden gäller vid pH 7. Vid lägre pH-värden får pE-gränserna justeras uppåt och vid högre pH-värden fär pE-gränserna justeras neråt.
Ibland används bara uttrycken oxiderande förhållanden och reducerande förhållanden, där oxiderande förhållanden råder vid höga pE-värden och reducerade förhållanden råder vid låga pE-värden.
pE-pH diagram
Då nästan alla redox-reaktioner i markvätskan påverkas av (och påverkar) dess pH-värde, är det ofta lämpligt att göra ett s.k. pE-pH-diagram för relevanta grundämnen. I pE-pH-diagrammet framgår tydligt vilka kemiska former av t.ex. järn, mangan eller svavel som dominerar vid en given kombination av pE och pH. Ofta plottas pH på x-axeln och pE på y-axeln.
Det matematiska sambandet mellan pE och pH
Om vi tänker oss den allmänna redox-reaktionen
- mAox + nH+ + e-
 pAred + qH20
pAred + qH20
så blir jämviktskonstanten (K)
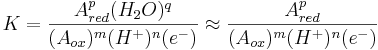
då vattenaktiviteten (H2O) brukar kunna sättas till 1. Om uttrycket för K logaritmieras, får vi
- log K = p*log (Ared) - m*log (Aox) - n* log (H+) - log (e-)
Då pH = -log (H+) och pE = -log (e-), får vi
- log K = p*log (Ared) - m*log (Aox) + n*pH + pE
var på pE kan lösas ut
- pE = log K + m*log (Aox) - p*log (Ared) - n*pH
Om både den reducerade formen (Ared) och den oxiderande formen (Aox) är vattenlösliga, inträffar jämviktsläget mellan de olika formerna när
- p*log (Ared) = m*log (Aox)
varpå sambandet mellan pE och pH kan förenklas till
- pE = log K - n*pH
Det är detta samband som ritas ut på pE-pH diagrammet.
Att mäta pE och pH i markvätskan
Både pE och pH mäts på liknande sätt, varför bägge mätningarna tas upp här.
Att mäta pE
Om redoxreaktionen sker i en vattenlösning, kan pE mätas med hjälp av elektroder. Man nyttjar då sambandet mellan pE och elektrodpotentialen:
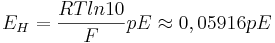
där
EH = Elektrodpotential (V)
R = Allmänna gaskonstanten (8,314 4 J/(K mol))
T = Temperatur (298 K)
F = Faradays konstant (96 486 C/mol)
Om man använder en ren platina-elektrod tillsammans med en kalomel-elektrod (kalomel = Hg2Cl2), så oxideras platinan, och kalomelen reduceras enligt följande halvreaktion:
- 1/2 Hg2Cl2(s) + e-
 Cl-(aq) + Hg*(l) (log K = 4,53).
Cl-(aq) + Hg*(l) (log K = 4,53).
Med tillämpning av termodynamikens lagar, kan man beräkna elektrodpotentialen mellan två elektroder kan genom formeln:
- E = B + Ej + EH = B + Ej + 0,05916(pE)
där
B = en parameter som beror på log K. B är unik för varje kalomelelektrod.
Ej = "vätskekopplingspotentialen" i en saltbrygga bestående av mättad lösning av kaliumklorid (som hindrar kloridjonerna från att diffundera ut från kalomel-elektroden). Ej beror på rådande jämviktförhållanden i saltbryggan.
Det sammanlagda värdet av B + Ej fås genom kalibrering av elektroderna mot olika redox-buffertar med kända egenskaper, t ex en blandning av 0,1 M järn(II)sulfat + 0,1 M Fe(III)sulfat i en 1 M svavelsyra.
Praktiska problem med pE-mätning i markvätskan
Det finns ett fleral praktiska problem med att mäta pE-värdet i markvätskan:
- Platinaelektroden svarar på flera halvreaktioner, utöver kalomel-elektroden.
- Koncentrationen av många redoxämnen är för låg i markvätskan, för att kunna påverka platinaelektroden.
- Vissa redoxämnen (t ex sådana ämnen som reducerar kol, kväve och svavel) är inte elektroaktiva.
- Platinaelektroden kontamineras av oxider och andra föroreningar.
- Ej i markvätskan skiljer sig rejält från Ej i redoxbufferten.
En pE-mätning ger bara ett kvalitativt mått på markens redoxpotential. Det uppmätta pE-värdet duger bara till att typkategorisera marken som oxisk, suboxisk eller anoxisk.
pH-mätning
Om kalomelelektroden behålls oförändrad, medan man byter ut pE-mätarens platinaelektrod mot en glasmembranelektrod, får man en pH-mätare. Glasmembranet fungerar som en adsorbtionsyta för markvätskans protoner. Den pH-skillnad som finns mellan markvätska och glasmembranets innervätska skapar en mätbar elektrodpotential. Sambandet mellan pH och elektrodpotentialen ser ut på följande sätt:
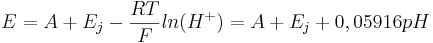
där
E = Elektrodpotential mellan två elektroder (V)
A = en parameter som beror på log K. A är unik för varje kalomel elektrod.
Ej = "vätskekopplingspotentialen" i en saltbrygga bestående av mättad lösning av kaliumklorid (som förhindrar kloridjonerna från att diffundera ut från kalomel-elektroden). Ej beror på de rådande steady-state förhållandet i saltbryggan.
R = Allmänna gaskonstanten (8,3144 J/(K mol))
T = Temperatur (298 K)
F = Faradays konstant (96486 C/mol)
(H+) = vätejons-aktiviteten
När det gäller proton-aktivitet, sker kalibreringen direkt mot buffertlösning med känt pH-värde. Därför mäts bara relativa pH-värden:
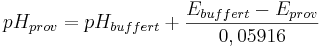
där
pHbuffert = pH-värdet i bufferten.
pHprov = pH-värdet i det undersökta provet (markvätskan)
Ebuffert = Elektrodpotentialen i bufferten (V)
Eprov = Elektrodpotentialen i det undersökta provet (V)
Praktiska problem med pH-mätningar av markvätskan
Det principiella problemet med pH-mätningar i markvätskan är osäkerheten i vätskekopplingspotentialen (Ej). Då markvätskans innehåll skiljer sig radikalt från pH-buffertens innehåll, så kommer vätskekopplingspotentialen att förändras. Det är omöjligt att förutse hur stor skillnaden kan tänkas bli. Blir skillnaden alltför stor, så blir det uppmätta pH-värdet oanvändbart för att kunna karakterisera det kemiska tillståndet i marken.
Källor
- Sposito, Garrison: The Chemistry of Soils, 277 sid. Oxford University press, New York 1989. ISBN 0-19-504615-3.